Psi4 + Antechamber进行Sigma Hole的RESP电荷拟合
分子力场中,为描述复杂分子体系的电荷分布,常常会引入带有电荷的哑原子进行描述。最经典的哑原子分子比如TIP4P和TIP5P的水分子力场,4点水模型TIP4P将O的负电荷在H-O-H的夹角内的哑原子上,O的质量则留在O原子处;5点水模型TIP5P则将O的负电荷拆分在O原子的Lone Pair处,模拟O的孤电子对。
除去水分子外,AmberTools的MDGX模块提供了添加哑原子,或虚位点(Virtual Site)的方法,如教程35中对氯苯的苯环上哑原子,及Sigma Hole哑原子添加的方法。不过MDGX使用较为复杂,MDGX的电荷拟合教程也较复杂。本文将从Antechamber的RESP电荷拟合讲起,讨论添加哑原子后直接使用Antechamber进行哑原子电荷拟合的方法。
分子力场中的Sigma Hole
生物体系模拟中的常用力场大部分不带有Sigma Hole的哑原子电荷,需要专业技术人员的手动添加和拟合。在CHARMM general force field (CGenFF) 于2016年发表的文章中,提出了一个非常简单的策略,即在Cl,Br,I的反键方向的固定距离上,添加+0.05的正电荷,从而在宏观模拟中的液体性质和分子间相互作用获得较大的提升。OPLS力场则有一系列文章讨论不同原子Sigma Hole的描述和影响。从原理上看,这一策略理应可以直接移植到GAFF力场中,也会有一个从无到有的提升。
但是固定电荷固定距离的方法从原理上是有瑕疵的,Br,I的原子半径更大,原子核对外层电子的吸引力更弱,因此成键后形成的Sigma Hole应该更大。卤素连接不同强弱的吸电子基团时,Sigma Hole的大小和位置也会发生变化。另外,噻唑或噻吩的S原子的S-C键的反键轨道上也存在Sigma Hole,而CGenFF中并没有添加此类Sigma Hole的拟合。因此本文提供一种操作简单且通用的Sigma Hole或Lone Pair的RESP电荷拟合流程。对此我们首先需要了解Antechamber中RESP电荷拟合的流程,并在这一流程中添加哑原子的坐标进行电荷拟合。
Antechamber做了什么
查阅AmberTools的官方文档,可以发现Antechamber本质是一个流程软件,会调用atomtype,bondtype,sqm,am1bcc,espgen,respgen,resp等等。其中atomtype指认GAFF力场的原子类型,bondtype指认GAFF的键类型,sqm和am1bcc进行AM1-BCC电荷计算,espgen读入量化软件输出转化为Antechamber ESP文件,respgen生成resp输入参数文件,resp则进行两步RESP电荷拟合。resp程序就是直接分发的RESP电荷提出者开发的软件。
衔接上篇文章中RESP计算,运行以下命令:
1 | antechamber -i gas.gesp -fi gesp -o gas.mol2 -fo mol2 -c resp -nc 0 |
我们可以看到一些中间文件,其中3个文件比较重要,ANTECHAMBER.ESP,ANTECHAMBER_RESP1.IN,ANTECHAMBER_RESP2.IN,分别是Antechamber的ESP文件,和RESP的Stage 1和Stage 2的RESP拟合输入参数文件。这三个文件都是resp程序进行拟合时的输入文件,关于三类文件的文档见RESP文档
首先是ANTECHAMBER.ESP:
1 | 5 2969 0 |
ANTECHAMBER.ESP文件格式就是精简版的GESP文件,单位均为原子单位AU。该文件中第一行共3列,第一列为原子数,第二列为ESP格点数,第三列为电荷值;第二行开始为原子坐标,每行3列;最后一部分为ESP格点,为4列,第1列为静电势,其余三列为格点坐标。
然后是ANTECHAMBER_RESP1.IN,即Stage 1的非烷烃H原子对称限制下的RESP拟合步:
1 | Resp charges for organic molecule |
具体文档见RESP文档,我们主要注意的是Resp charges for organic molecule后的内容。这一部分第一行共2列,第一列为Net Charge,第二列为原子数,每列宽度为5。接下来N行为原子的元素和组编码(ivary),组编码用于对称性限制。这里的对称限制指等价原子电荷应当相等,如亚甲基CH2的两个H化学环境相同,一般应该带有相等的电荷才复合实际模拟情况。本文例子CF3Cl中,三个F原子化学环境等价,应该限制为相等电荷。因此参数文件中的第3、4号原子(F原子)信息 9 2写入的组编码都为2,即与2号原子(F原子)电荷一致。其余组编码为0的原子则代表不启用对称性限制。
最后是ANTECHAMBER_RESP2.IN,即Stage 2的烷烃H原子对称限制下的RESP拟合步:
1 | Resp charges for organic molecule |
第二步拟合时没有对称限制,因此组编码均为-99/-1时即不启用,且固定电荷在拟合中不再变化。所以事实上这一步运行前后不会对电荷进行任何拟合。注意:RESP电荷拟合原文中明确指出第一步是完全非对称限制的,允许原子电荷最大程度自由拟合,第二步是其他原子不再拟合,对sp3杂化的C原子拟合,以及对应的氢加对称限制拟合。但是上文中提及的Antechamber中的运行步骤是第一步直接使用对称限制,第二步大部分原子电荷直接固定,少量原子再进行对称或非对称拟合,其中原理阅读Antechamber中生成RESP参数的respgen.c源码可以得出大致结论:RESP Stage1对非与C相连的H原子以外其余对称原子也添加对称限制,RESP Stage2则与论文一致,将sp3杂化的C和与C相连的对称H原子添加对称性质的同时固定其他电荷。CF3Cl中没有连接对称H原子的sp3杂化C,因此RESP Stage2的组编码均为-99。目前我们仍然以Antechamber的中间文件作为参考进行后续修改以保持与官方的拟合一致性。
基于以上文档,我们可以明白,只需要在ESP文件中添加一行哑原子坐标,在RESP的两个参数文件添加一行哑原子的的元素类型(0)和组编码即可完成对Sigma Hole哑原子的电荷拟合。
简单手动添加哑原子的过程
仍然使用上篇文章中Psi4 RESP计算样例CF3Cl,运行以下命令:
1 | antechamber -i gas.gesp -fi gesp -o gas.mol2 -fo mol2 -c resp -nc 0 |
获得ANTECHAMBER.ESP,ANTECHAMBER_RESP1.IN,ANTECHAMBER_RESP2.IN三个文件,然后在ESP中添加一行哑原子坐标,坐标位置我们使用CGenFF中的Cl元素Sigma Hole的位置,即Cl-C键反键方向1.64 Å,这里使用原子单位,转换为Bohr为3.157 Bohr,笛卡尔坐标系下为-1.6365765E+00 1.2314168E+00 1.7220899E+00。修改后ANTECHAMBER.ESP变为下边的样子。
1 | 6 2969 0 |
然后在ANTECHAMBER_RESP1.IN和ANTECHAMBER_RESP2.IN也添加哑原子的元素ID(0)和组编码信息
1 | Resp charges for organic molecule |
1 | Resp charges for organic molecule |
最后调用两次resp命令,即可获得
1 | mkdir MOD1; mkdir MOD2 |
qout文件中的电荷
1 | 0.540036 -0.141469 -0.141469 -0.141469 -0.176623 0.060993 |
最后一个电荷为虚原子电荷,为+0.0609,比CGenFF中的+0.05略大,也比后文氯苯中的+0.0550略大,符合CF3Cl中F原子的强拉电子作用的预期影响。
进一步将点电荷的静电势进行可视化复现 (Figure 2),可以看到没有Sigma Hole哑原子的ffESP情况下,CF3Cl的整体静电势趋于低估,无法体现Sigma Hole效应和足够的偶极矩。在添加Sigma Hole的ffESPSH情况下,体现出了Sigma Hole效应和偶极矩。从数据角度看,添加Sigma Hole后,分子的偶极从0.371 Debye增加到0.440 Debye,非常接近量化计算的0.442 Debye;ESP拟合的relative RMS从0.76降低到0.31,也更接近量化计算的ESP。

Figure 1. RESP Model for CF3Cl With or Without Sigma Hole
结论
本文介绍了一种简单修改Antechamber中间文件进行Sigma Hole电荷拟合的方法,该方法也可以扩展到其他哑原子电荷拟合中。除本文例子中的CF3Cl分子外,我还测试了其他分子,如噻吩的S原子Sigma Hole电荷为0.026,碘苯的I原子Sigma Hole电荷为0.065,与化学经验和量化计算结果吻合。但是邻位酰胺取代的噻吩的S原子Sigma Hole电荷为0.016,这显然是因为S与酰胺的O形成氢键后,一侧的Sigma Hole不明显,强行对称化S的一对Sigma Hol导致的低估。尽管如此,这一做法仍然比没有Sigma Hole时更好地描述了邻位酰胺取代的噻吩的最低能量构象。这一问题单一构象RESP一定无法解决,需要多构象RESP拟合,但是此例低能量构象占权重较大时,这一问题仍然存在,希望读者提供一些建议。
本文中并没有讨论合理的Sigma Hole的反键距离多少合适,这是一个多参数的优化问题。考虑到计算成本的问题,使用CGenFF的距离是一种简便方法。另外,本文也没有写明如何将虚原子添加在Amber力场文件中,我这里提供一份CF3Cl分子的Gromacs ITP文件的哑原子描述方法,即使用Gromacs的[ virtual_sites2 ]模式添加反键方向的哑原子,供参考。
1 | ; opt_GMX.itp created by acpype (v: 2023.10.27) on Wed Feb 14 23:50:46 2024 |
补充 20240219
补充几个化合物的计算样例。
样例1. 芳香化合物的Sigma Hole
对氯苯,溴苯和碘苯以及噻吩和噻唑的Sigma Hole进行RESP拟合。
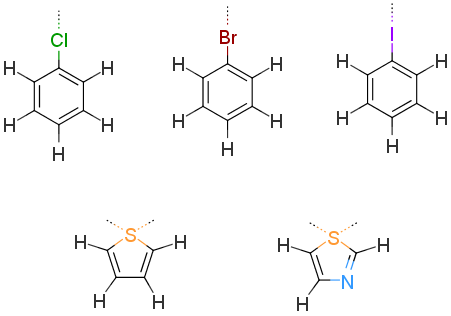
Figure 2. RESP Model Systems for Sigma Hole
不同方法计算后的RESP(m06-2x-d3zero2b/def2-tzvp)电荷和CGenFF电荷对比。
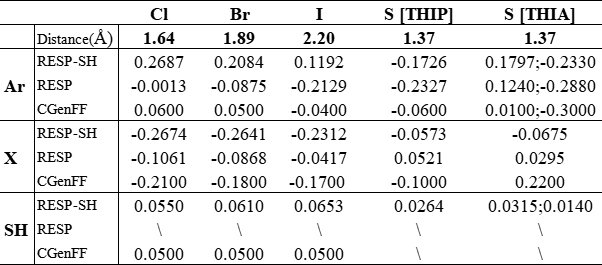
Table 1. RESP Model Systems for Sigma Hole
根据表1,Cl, Br, I的Sigma Hole所带的正电荷依次增加。噻唑对比噻吩,S的两侧化学环境是非对称的,左侧的Sigma Hole由于间位N的吸电子效应,从0.0264增加到0.0315,绝对值变大;右侧的Sigma Hole从0.0264减小到0.0140,变小。以上计算结果与化学家的化学直觉和实验观测是较为一致的。
样例2. 2-乙酰基噻吩的最低能量构象
2-乙酰基噻吩有一个可旋转键,因为羰基与噻吩环共轭,所以理论上会有两个能量较低的构象,即羰基和S同侧或异侧。
Figure 3. 2-乙酰基噻吩
根据量化(B3LYPD3/6-311G*)对2-乙酰基噻吩的二面角进行势能面扫描,如Figure 3所示,两个局部能量极小点中,S和羰基同侧的构象为全局能量极小点,相比S和羰基对侧能量低1.8 kcal/mol。可以从静电势角度进行解释,即噻唑S的Sigma Hole带有正电,与羰基氧同侧时能够形成静电吸引,从而降低了同侧构象的自由能。因此,含有Sigma Hole的力场模型才有可能正确描述这一构象分布情况。
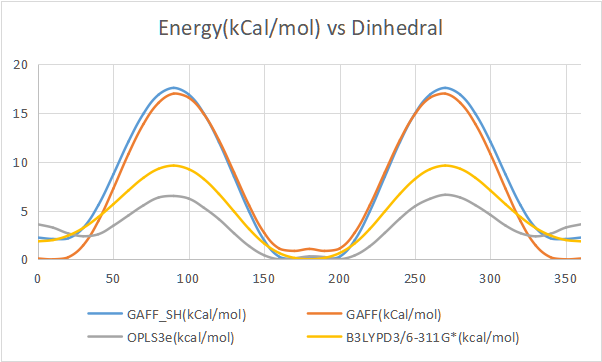
Figure 4. 2-乙酰基噻吩不同力场或电荷模型下二面角势能面扫描
对此我们也通过对GAFF, GAFF+Sigma Hole (GAFF_SH)及OPLS3e力场分别进行2-乙酰基噻吩的二面角势能面扫描。没有Sigma Hole的GAFF力场给出了完全相反的能量排序,认为S和羰基氧同侧时能量更高,这显然是不合理的。添加Sigam Hole电荷描述后,GAFF_SH给出了最接近量化计算的能量差,~1.8 kcal/mol;但是与GAFF类似,势能面整体表现较差。OPLS3e也很好的描述了S和羰基氧同侧时能量最低,同时它的自由能面整体上比GAFF更接近量化计算结果,不会出现阻转异构现象。后续进一步分析认为是GAFF的二面角参数过大导致势能面能垒过高,产生了阻转异构,这会导致长时间模拟下分子无法发生翻转。因此,单纯修改电荷模型仍然不足,需要同时修改GAFF的二面角参数,来提高分子模拟的准确性。
Update:2024年初上海药物所在JMC(10.1021/acs.jmedchem.3c02359)的文章中探讨了Halogen Bond在蛋白-多肽结合的稳定性作用,其中Figure 4通过在HF/6-31G(d)(I使用LanL2DZ基组)条件下计算静电势,再使用ESP拟合方法对 卤素-SigmaHole 距离进行扫描,得到了Br-SH最优距离1.37Å,I-SH最优距离1.73Å。
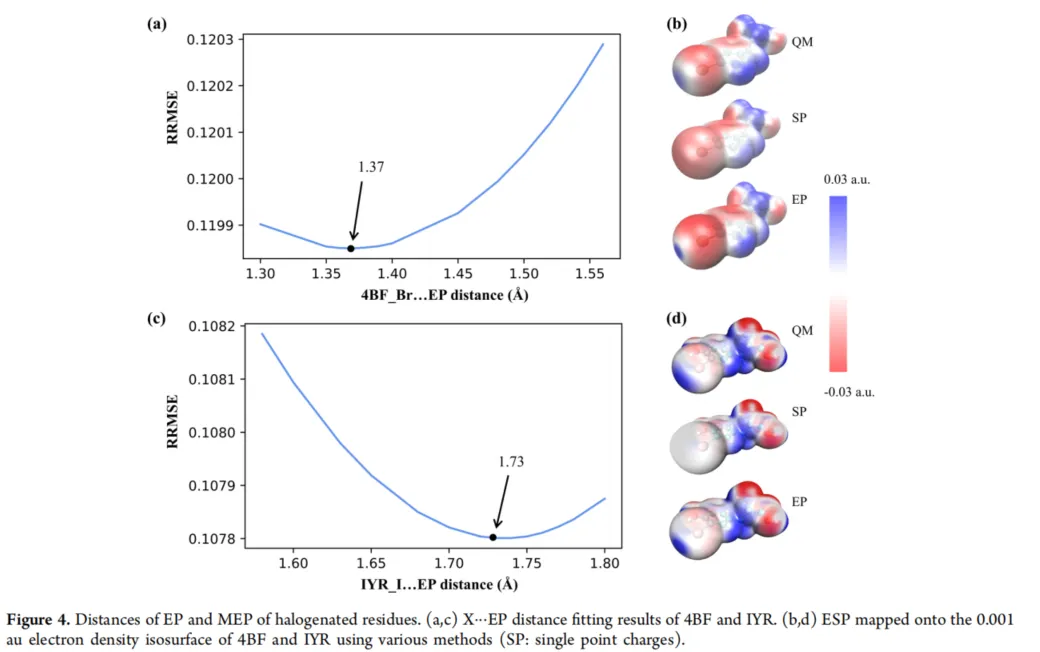
Figure 5. HF/6-31G(d)精度下的ESP拟合X···SH距离势能面扫描
参考文献
[1] https://ambermd.org/tutorials/advanced/tutorial35/index.php Tutorial 35
[2] https://ambermd.org/tutorials/advanced/tutorial28/index.php Tutorial 28
[3] Ignacio Soteras Gutiérrez b, et al. Parametrization of halogen bonds in the charmm general force field: improved treatment of ligand–protein interactions. Bioorganic & Medicinal Chemistry (2016), 24(20), 4812-4825.
[4] Jorgensen, et al. Improved Description of Sulfur Charge Anisotropy in OPLS Force Fields: Model Development and Parameterization. Journal of Physical Chemistry B Condensed Matter Materials Surfaces Interfaces & Biophysical (2017).
[5] Jorgensen, W. L., & Schyman, P. (2012). Treatment of Halogen Bonding in the OPLS-AA Force Field; Application to Potent Anti-HIV Agents. Journal of chemical theory and computation, 8(10), 3895–3801.
[6] https://ambermd.org/Manuals.php Amber Reference Manuals
[7] https://upjv.q4md-forcefieldtools.org/RED/resp/ RESP version 2.2
[8] Jintian Li, et al. (2024). mpact of Halogen Bonds on Protein–Peptide Binding and Protein Structural Stability Revealed by Computational Approaches. Journal of Medicical Chemistry, 67(6) 4782–4792