RESP电荷用于分子模拟,也是Amber/GAFF力场的御用电荷。Amber官方的的电荷教程以商业软件Gaussian和Jaguar为主,但目前使用开源的Psi4的Antechamber拟合教程并没有先例。除此以外,Multiwfn也是一种选择,Multiwfn支持多种量化格式的RESP和RESP2电荷的拟合。本文中,我们仅依靠python开发环境的Psi4和Antechamber进行有机小分子的RESP电荷拟合,以避开昂贵商用的Gaussian和Jaguar等软件.
Psi4作为基于python的开源量化软件,使用conda安装简单快捷。Ambertools开源后也通过anaconda进行分发,使用conda同样可以简单快速地安装。
1 conda install psi4 ambertools -c conda-forge
使用高斯计算配体的表面静电势(Electro Potential Surface, ESP)的过程详见此文 的解释。
Antechamber可以使用高斯的两种输出格式进行RESP拟合。第一种方式是使用高斯的output文件进行拟合,第二种方式则是使用高斯输出的GESP文件进行拟合。我们需要关注的是Antechamber使用GESP格式进行RESP的拟合命令,因为GESP文件的格式清晰简单,而且我们可以使用Psi4输出的静电势格点进行“伪装”。
1 2 3 4 5 6 7 antechamber -i ligand.gesp -fi gesp -o ligand_resp.mol2 -fo mol2 -c resp
让我们以分子CF3Cl为例进行计算,首先打开一个GESP文件解读下它的格式。GESP文件的生成见前文引用 。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 ESP FILE - ATOMIC UNITS CHARGE = 0 - MULTIPLICITY = 1 ATOMIC COORDINATES AND ESP CHARGES. #ATOMS = 5 C -0.66076449D+00 0.36262007D-03 0.47985687D-16 0.36304379D+00 F -0.15151018D+01 -0.11583839D+01 -0.20049910D+01 -0.10253982D+00 F -0.15151019D+01 -0.11584051D+01 0.20049788D+01 -0.10253982D+00 F -0.15165984D+01 0.23157629D+01 0.12246920D-04 -0.10187755D+00 Cl 0.26403415D+01 0.41522919D-03 0.13662726D-15 -0.56086592D-01 DIPOLE MOMENT: X= 0.10644740D+00 Y= 0.46159385D-03 Z= -0.48794012D-08 Total= 0.10644840D+00 TRACELESS QUADRUPOLE MOMENT: XX= 0.77021461D+00 YY= -0.38540685D+00 ZZ= -0.38480776D+00 XY= -0.47956518D-03 XZ= -0.13940940D-06 YZ= 0.75005058D-08 ESP VALUES AND GRID POINT COORDINATES. #POINTS = 3558 0.18293171D-01 -0.45329677D+01 0.39412720D+00 0.77420129D+00 0.20596410D-01 -0.46291894D+01 0.36262007D-03 0.29097360D-15 0.18734720D-01 -0.45529372D+01 -0.77383867D+00 0.29097360D-15 0.20696154D-01 0.11343680D+00 -0.38918101D+01 0.29097360D-15 0.18293167D-01 -0.45329677D+01 0.39412720D+00 -0.77420129D+00 .... Another 3553 Lines, 3558 Lines in total
高斯的GESP文件格式还是比较清晰的,在不查询文档时也可以通过文件内的区块名称进行理解。第一部分,ESP FILE区块指明了接下文件中所有单位为原子单位 (ATOMIC UNITS),即距离/坐标单位为Bohr (1 Bohr = 0.529 Å),静电势单位为E h /e,偶极单位为 ea 0
第二部分则是电荷CHARGE和自旋多重度MULTIPLICITY,这两个参数Antechamber不会读入,所以可以固定不变。具体的分子电荷通过Antechamber的-nc参数进行指定即可覆盖文件中所有电荷信息,或第三部分的偏电荷加和确定。
第三部分 ATOMIC COORDINATES AND ESP CHARGES为重点部分,即元素类型、原子坐标和原子的偏电荷,一共1+3+1列,行数即原子数,由#ATOMS指出。数字格式为科学计数法(D为Fortran语言习惯,使用e或E不会影响Antechamber读入),单位均为前文提到的AU。第5列的偏电荷可以留空,后续使用Antechamber的-nc参数进行指定即可覆盖。如果不留空,也不指定Antechamber的-nc参数时,Antechamber会将此列电荷加和作为Net Charge。
第四部分DIPOLE MOMENT和第五部分TRACELESS QUADRUPOLE MOMENT,即偶极矩和四极矩,Antechamber不会使用这几行数据,留下对应数量空行即可。
第五部分 ESP VALUES AND GRID POINT COORDINATES为重点部分,即静电势,静电势坐标,共1+3列,行数为静电势表面格点数,由#POINTS指定。所有数值为科学计数法,原子单位。
接下来的任务就很明确,首先使用Psi4计算表面静电势格点文件,然后将分子坐标,静电势格点坐标和静电势进行相应替换,即可以“伪装”高斯GESP文件,用于Antechamber的RESP拟合
Psi4单点优化过程与一般量化软件无异,设置坐标,提供量化方法和相应基组即可。这里我们参考sobereva文章 和这篇文章 ,使用m06-2x-d3zero2b/def2-svp进行构象优化。主要选择原因是添加D3校正后,M062X-D3在有机小分子中表现较好,def2系列基组则对超过第四周期的元素有更方便的支持。Psi4下的坐标单位都是Å,静电势单位为AU
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 # psi4_opt.in # Any line starting with the # character is a comment line memory 2 GB molecule CONF { 0 1 C 0.17930 0.30890 0.22760 F 0.55040 1.19750 -0.77480 F -0.47570 -0.77930 -0.33700 F 1.31350 -0.13090 0.90010 Cl -0.88500 1.10940 1.34740 } set { basis def2-svp freeze_core True } optimize('m06-2x-d3zero2b') CONF.save_xyz_file("opt.xyz", 1)
然后运行
1 psi4 psi4_opt.in opt.out -n 20
最后一行导出opt.xyz作为优化后结构,由于后续单点能和静电势计算。
高斯或其他量化软件的静电势可以通过GaussView或Mutiwfn进行可视化分析,导出.cube文件后,可以借助VMD进行定制化的显示。不过我们这里不使用.cube文件,而是通过E, wfn = prop('m06-2x-d3zero2b', properties=["GRID_ESP", "GRID_FIELD"], return_wfn=True) 函数直接导出类似GESP文件的静电势格点。
在进行计算前,我们先溯源Amber力场的RESP用的格点是如何指定的。RESP电荷使用的格点坐标是通过在分子的原子范德华表面壳层进行采样获得的,为了平衡精度和计算量,RESP原文 中使用了1.4, 1.6, 1.8, 2.0倍的VDW半径的壳层,并且要求每平方埃至少采样1个点。不同的程序和采样方法在这里会引入一些差异,比如在球面上的拟合的算法是否相同,在原子接缝的地方是否自动去除过密的拟合点,密度定义是体积还是面积等。不过此类差异在采样点数量接近时,对最后的电荷拟合产生的影响可以忽略不记。
Github中Psi4的RESP已经有一些实现,因此我们可以直接借鉴CSDGroup的范德华表面采样代码 。我们可以根据自身的需求对代码进行微调。
比如源码中,原子VDW半径取自GAMESS,只有前3个周期。如果我们想拟合有机小分子中也有的Br和I元素的话,需要手动添加对应原子的VDW半径。一种简单方法是直接使用全周期的UFF力场中的原子半径除1.2进行近似。
1 2 3 4 5 6 vdw_r = {'H' : 1.20 , 'HE' : 1.20 , 'LI' : 1.37 , 'BE' : 1.45 , 'B' : 1.45 , 'C' : 1.50 , 'N' : 1.50 , 'O' : 1.40 , 'F' : 1.35 , 'NE' : 1.30 , 'NA' : 1.57 , 'MG' : 1.36 , 'AL' : 1.24 , 'SI' : 1.17 , 'P' : 1.80 , 'S' : 1.75 , 'CL' : 1.70 }
核心代码为
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 def vdw_surface (coordinates, elements, scale_factor, density, input_radii ): """Computes points outside the van der Waals surface of molecules. Parameters ---------- coordinates : ndarray cartesian coordinates of the nuclei, in units of angstrom elements : list The symbols (e.g. C, H) for the atoms scale_factor : float The points on the molecular surface are set at a distance of scale_factor * vdw_radius away from each of the atoms. density : float The (approximate) number of points to generate per square angstrom of surface area. 1.0 is the default recommended by Kollman & Singh. input_radii : dict dictionary of user's defined VDW radii Returns ------- radii : dict A dictionary of scaled VDW radii surface_points : ndarray array of the coordinates of the points on the surface """ ... for i in range (len (coordinates)): n_points = int (density * 4.0 * np.pi* np.power(radii[elements[i]], 2 )) dots = surface(n_points) ... return np.array(surface_points), radii def surface (n ): """Computes approximately n points on unit sphere. Code adapted from GAMESS. Parameters ---------- n : int approximate number of requested surface points Returns ------- ndarray numpy array of xyz coordinates of surface points """ ... return np.array(u)
摘出的代码即对原子球表面进行格点采样的核心代码,具体的代码实现和使用请到Github的vdw_surface.pydriver.py
最后我们将算出的分子VDW表面导出为gird.dat格点文件即可np.savetxt('grid.dat', points, fmt='%15.10f')。这个文件放置在接下来的Psi4单点能计算的同一目录下,Psi4会自动调用并计算grid.dat坐标对应的静电势。
接下来计算交给Psi4
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 # psi4_gas.in # Any line starting with the # character is a comment line memory 2 GB molecule CONF { 0 1 C 0.210029298380 -0.158090055990 -0.221115071776 F 0.570509626472 0.672705716575 -1.177444086426 F -0.397910302325 -1.192163020236 -0.764308861674 F 1.290496394679 -0.580428741997 0.402797484019 Cl -0.866966788650 0.651813183796 0.911985231755 } set { basis def2-tzvp freeze_core True } # set cubeprop_tasks ['esp'] # 与cubreprop函数配合输出静电势的.cube文件,用于可视化,RESP计算不需要 # psi4 会自动调用grid.dat文件计算properties=["GRID_ESP",] E, wfn = prop('m06-2x-d3zero2b', properties=["GRID_ESP",], return_wfn=True) # cubeprop(wfn)
1 psi4 psi4_gas.in gas.out -n 20
运行顺利时,计算目录下会写出grid_esp_gas.dat文件,该文件仅有一列数据,即静电势,单位为AU。
这一步几乎不需要计算,只需要进行单位和格式转换即可。坐标值单位需要转换为Bohr,静电势单位无需转换,其余位置留空或修改即可。此步使用python脚本进行即可。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 ESP FILE - ATOMIC UNIT CHARGE = 0 - MULTIPLICITY = 1 ATOMIC COORDINATES AND ESP CHARGES. #ATOMS = 5 C 3.96897854e-01 -2.98746910e-01 -4.17846930e-01 0.00000000e+00 F 1.07810695e+00 1.27122957e+00 -2.22504686e+00 0.00000000e+00 F -7.51941497e-01 -2.25286161e+00 -1.44433443e+00 0.00000000e+00 F 2.43868476e+00 -1.09685136e+00 7.61176932e-01 0.00000000e+00 Cl -1.63832980e+00 1.23174841e+00 1.72340233e+00 0.00000000e+00 DIPOLE MOMENT: TRACELESS QUADRUPOLE MOMENT: ESP VALUES AND GRID POINT COORDINATES. #POINTS = 2969 1.78249666e-02 -4.46537858e-02 -3.36980602e+00 2.05642551e+00 1.85040189e-02 -2.58006295e+00 5.75367651e-01 -2.89211937e+00 1.86787356e-02 3.00700430e+00 -1.97616047e+00 -2.89211937e+00 1.71942816e-02 2.29230051e+00 -1.88917858e+00 -3.52048644e+00 1.52906452e-02 2.82837361e+00 2.86680845e+00 4.48320948e-01 .... Another 2964 Lines, 2969 Lines in total
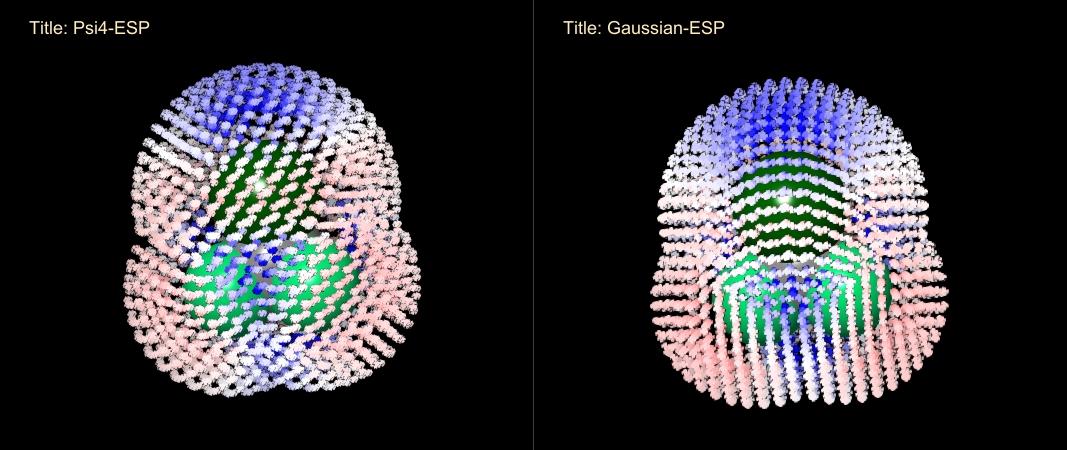
这里将两个GESP文件分别可视化,对静电势以-0.01~+0.01进行着色,可以看到CF3Cl分子的表面静电势在Psi4和高斯中计算结果基本一致,且Cl的Sigma Hole的正静电势也清晰可见。
接下来的流程与官方文档一致,额外需要说明的时尽量使用-nc参数指定Net Charge,以覆盖“伪造”的GESP文件中的电荷。
1 2 3 antechamber -i gas.gesp -fi gesp -o ligand_resp.mol2 -fo mol2 -c resp -nc 0
到此,Psi4+Antechamber的RESP电荷拟合完成。本教程尽量使用最少的依赖,只使用基于Conda分发的Psi4和AmberTools及一些辅助脚本,即可完成RESP电荷的拟合,告别对商业软件高斯的依赖。以上代码后续会发布在我的github中,供读者使用和调整。
思考题 :CF3Cl由于F的拉电子作用和Cl本身的性质,在C-Cl键的反键轨道处产生Sigma Hole,即此区域的静电势为正。前文中RESP电荷拟合后Cl带-0.049的负电荷,是无法体现正电区域的。假如我们想引入一个带正电荷的哑原子进行RESP,该如何达成?
[1] http://gohom.win/2015/09/17/AutoCalcRESP/ 自动计算ESP和RESP电荷(AMBER and G09)
[2] http://sobereva.com/441 RESP拟合静电势电荷的原理以及在Multiwfn中的计算
[3] http://sobereva.com/336 谈谈量子化学中基组的选择
[4] Bayly, C. I., Cieplak, P., Cornell, W. D. , & Kollman, P. A… (1993). A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the resp model . Journal of Physical Chemistry, 97(40), 10269-10280.
[5] https://github.com/cdsgroup/resp/blob/master/resp/vdw_surface.py vdw_surface.py