Shape Similarity and Electroshape Similarity Calculation
Now I want to introduce Molecular Shape Comparison function of Open Drug Discovery Toolkit (oddt). First of all, 3D structure of molecular is required for the calculation, and I use Maestro to generate MOL2 file of example moleculars.
USR (Ultrafast Shape Recognition) - function usr(molecule)
Ballester PJ, Richards WG (2007). Ultrafast shape recognition to search compound databases for similar molecular shapes. Journal of computational chemistry, 28(10):1711-23. http://dx.doi.org/10.1002/jcc.20681
USRCAT (USR with Credo Atom Types) - function usr_cat(molecule)
Adrian M Schreyer, Tom Blundell (2012). USRCAT: real-time ultrafast shape recognition with pharmacophoric constraints. Journal of Cheminformatics, 2012 4:27. http://dx.doi.org/10.1186/1758-2946-4-27
Electroshape - function electroshape(molecule)
Armstrong, M. S. et al. ElectroShape: fast molecular similarity calculations incorporating shape, chirality and electrostatics. J Comput Aided Mol Des 24, 789-801 (2010). http://dx.doi.org/doi:10.1007/s10822-010-9374-0
Calculate electro shape of moleculars
Then we should import oddt package and read file into python. toolkit.readfile returns a generator, so that we used next function to import molecular, and calculated electro shape. All compounds are co-crystalized ligands of Human Smoothened receptor (SMO), you can find them on RCSB PDB. I extracted their 3D coordinates from PDB file and saved as MOL2 file.
1 | from oddt import toolkit |
Then we use shape.usr_similarity calculate similarity of each pair of compounds.
1 | import matplotlib.pyplot as plt |
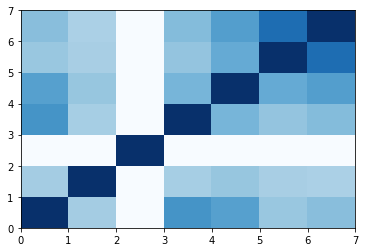
Conclusion
Obviously, LY2940680 is the most unique ligands amoug all this structure. All compounds, except Cholestrol and 20(S)-OHC, are different to each other. This is a result of selection of crystalization. Another key conclusion is that Cyclopamine is similar with Cholectrol and 20(S)-OHC (Hydroxy-Cholestrol), which is in accordence with the biochemical and crystal evidence.
USR and USR_CAT calculation
Next, similar result for other comparison funtion:
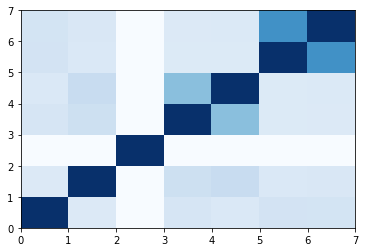
USR
1 | sant1 = shape.usr(next(toolkit.readfile('sdf', 'mol/sant1.sdf'))) |
USR_CAT
1 | sant1 = shape.usr_cat(next(toolkit.readfile('sdf', 'mol/sant1.sdf'))) |
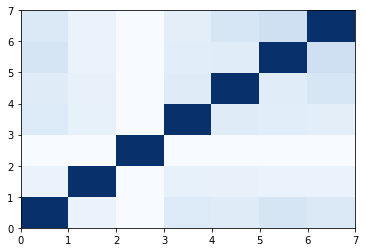
We have same result like electro shape similarity. Althogh you can use tanimoto similarity for 2D molecular, I would still recommend eclectroshape comparison function in oddt as an option.