Use Python to Generate Protein Contact Map of Molecular Dynamics
Protein contact map is a very helpfull tool to represent 3D protein structure in a 2D matrix format. More detail about contact map can be found here. Contact maps can not only be used as illustrations of protein, but also as tools to predict homology protein structure, especially for low homolog in evolution. Some scientists have already applied deep learning on this project and developed programs for structure prediction, such as RaptorX of TTIC.
There are several tools or program for calculate and display contact map on internet, here is an collection of them. I had a project several month ago, and I wanted to use a script to generate contact maps for a molecular dynamic trajectory, so that I use python to write a script to do it. I love python because I could always find usefull packages from internet. Here I found a package named MDtraj, And a package from Benjamin Rafferty (doi:10.4231/D35M62761)! Thanks to them, I can simply achieve my idea.
Calculate electro shape of moleculars
I used a protein folding trajectory from internet. I will add the internet address after I update this blog next time. 😛
1 | import mdtraj as md |
I use gromacs format here because Gromacs is an advaced and open software of molecular dynamics, although MDtraj supports plenty of trajectory formats.
1 | traj = md.load(XTC, top = PDB) |
1 | Frames: 401 Atoms: 287 Dimentions: 3 |
It is pretty convient to use MDtraj select residues and atoms.
hape Similarity and Electroshape Similarity Calculation
1 | topology = traj.topology |
1 | MD imformation: <mdtraj.Topology with 1 chains, 35 residues, 287 atoms, 290 bonds> |
Function to Calculate Cotact Map
Below are functions for contact map calculation. I modified source code obtained from: https://nanohub.org/resources/contactmaps
1 | def calc_residue_dist(residues = (0, 0)): |
Function for Output
Define functions for display and output of contact map. Also modified from https://nanohub.org/resources/contactmaps
1 | def draw(matrix, fig=None, cmap = 'binary', interpolation = 'nearest', colorbar = True, contour = False): |
Generate conact maps for a trajectroy
1 | for i in range(0, 400, 40): |
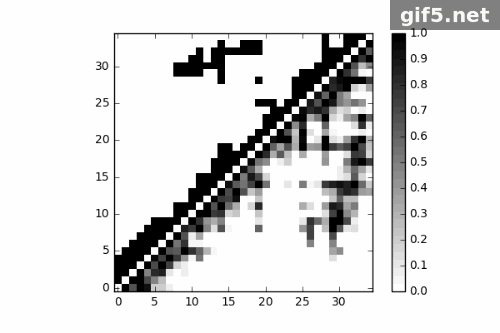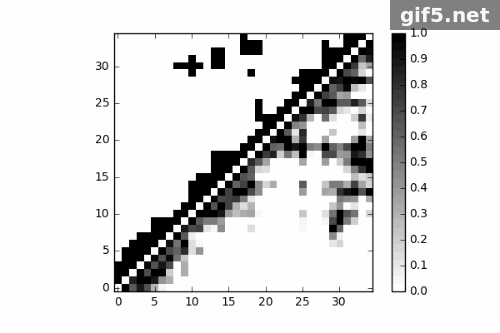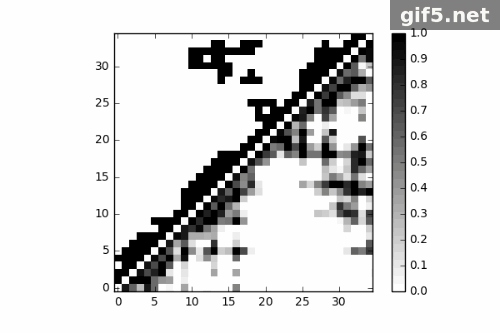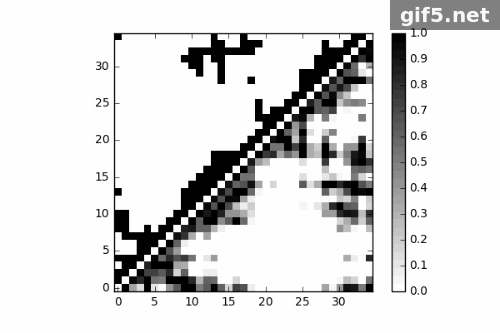
Conclusion
Finally, I generate a Gif picture for 11 frames of protein trajectory. The script could also generate matrix data of contact map, what can we do to that? ;P
References
[1] McGibbon, Robert T., et al. “MDTraj: A modern open library for the analysis of molecular dynamics trajectories.” Biophysical journal 109.8 (2015): 1528-1532.
[2] Benjamin Rafferty; Zachary Carl Flohr; Ashlie Martini (2014), “Protein Contact Maps,” https://nanohub.org/resources/contactmaps. (DOI: 10.4231/D35M62761).