论文笔记:药物de novo设计与深度增强学习
最近拜读了一篇来自AstraZeneca R&D的深度学习结合化学信息学的文章,Molecular De-Novo Design through Deep Reinforcement Learning。在这篇文章中,作者Marcus Olivecrona使用了RNN的方法进行了inverse QSAR,从而达到设计新的活性药物分子。而且作者的方法中可以通过inforcement learning增加各种限制,从而达到精准的调控。作者的Github项目地址在这里。由于文章比较复杂,因此在这里稍作笔记以深入学习。
1. Introduction
传统方法的药物筛选的空间可达1060 ~ 10100的可合成分子。
1.1. 经典 de novo 设计方法包括:
- 从化学空间和带电性上进行增长。 缺点:产生的分子具有较差的药代动力学(DMPK)性质
- 有机合成专家设计同系物。缺点:思维受限
- 反向定量构效关系Inverse QSAR:从活性数据产生QSAR,再将QSAR反向生成结构组合。缺点:QSAR的descriptor可能不可逆,难以反向生成结构。
1.2. 卷积神经网络RNN:
RNN通常被用于连续性数据(Sequencial Data)的建模,比如自然语言处理NLP,或音乐的自动编程。Segler等人证明,RNN在canonical SMILES的模型训练中,可以正确的学习到它的语法和化学空间分布;他们也在进一步的训练中证明了微调(fine-tuned)的模型可以预测大部分的活性。另外,有最近的两篇文章也指出增强学习reinforcement learning方法可以fine tune预训练的RNN模型。
2. Method
2.1. Recurrent Neural Network
RNN可以利用步骤之间的对称性的同时保持追踪之前步骤中,可能影响当前步骤的,最突出信息。它通过引入___cell___概念达成这一目的。对任意步骤___t___,___cellt___是前一个___cellt-1___和当前输入___xt-1___的输出结果。___cellt_的内容即会影响当前的输出,也会影响下一步下一个cell状态。因此network就有了对过去状态的__记忆,用以处理当前的新数据。因此RNN很适合做自然语言处理,在这一前提下,一个词组序列就可以编码为one-hot vector X。两个额外的tokens,GO,___EOS___被用来指代序列的开头和结尾。
2.1.1 Learning the data
训练RNN的方法同常使用极大似然估计,如,在给予前一步token的情况下,最大化在目标序列中下一个token ___xt___的log极大似然度。每一步模型都会生成一个基于下一个可能的词character的概率分布函数,而目标就是最大化对应正确token的极大似然度:
J(Θ) = −∑Tt=1 logP(xt∣xt−1,…,x1)
损失函数cost function L(Θ),通常作用于一个训练样本的子集,即batch,并且基于神经网络的参数进行最小化。给出步骤t的一个预测的likelihood log P,对应于Θ的预测的梯度,被用来更新Θ。这一方法即back-propagation。另外,因为RNN中参数的改变,不仅仅影响当前时间_t_下的直接输出,也会迭代地影响前一个cell输出当前cell的信息流。这一多米诺现象在RNN中被称作back-propagation throught time(BPTT)。
由于BPTT的处理中会出现多次自乘,所以有可能会导致梯度爆炸或梯度消失。因此Hochreiter等人提出了__Long-Short-Term Memory cell (LSTM)__,即可以在信息流中更可控地决定哪些信息被保留,而哪些信息被丢失。__Gated Recurrent Unit (GRU)__就是一种简单的可以实现减少计算消耗和实现大部分能效的LSTM架构。
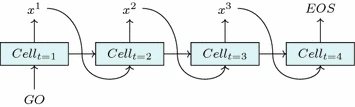
Figure 2. Sequence generation by a trained RNN
2.1.2 Generating new samples
一旦RNN被训练适用于目标序列后,它就可以根据训练集学习中产生的__条件概率分布__生成新的序列。第一个给出的input是___GO___ token,然后在我们每次根据预测的概率分布___P(Xt)___对output token ___xt___采样后,同时使用___xt___作为下一步的input。
2.1.3 Tokenizing and one-hot encoding SMILES
SMILES被用于以字符串来表示分子结构的方法。SMILES在tokenize过程中,单字符原子的单个字符被视为一个token,双字符原子的两个字符(如,Br、Cl)被视为一个token,特殊的环境结构(如,咪唑的[nH])也被视为一个token。根据这一规则,在训练集中总计有86种不同的tokens。Figure 3 举例了分子表示为SMILES和one-hot vector的情况。
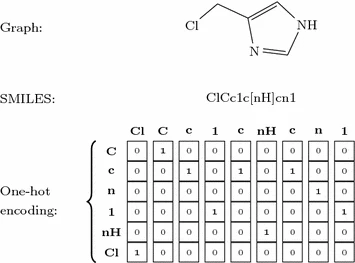
Figure 3. Three representations of 4-(chloromethyl)-1H-imidazole.
Three representations of 4-(chloromethyl)-1H-imidazole.
2.2. Reinforcement Learning
考虑一个Agent,对于某个状态_s_属于__S__,必须选择行动_a_属于__A__;这里__S__是可能状态的集合,__A__是相应状态的行动的集合。一个Agent的策略__policy π(a|s)将每一个状态state的概率匹配到每一个行动。许多增强学习框架被认为是Markov决策过程,即指当前状态包含所有用于决策的信息,而不需要了解之前的状态的历史。我们可以将这个概念一般化为部分观测的Markov决策过程,Agent可以与环境的不完整展示作用。另__r(a|s)为在某一状态下采取某一行动的优秀程度的回馈__reward,长期回馈 G(at, St = ∑Tt rt)__代表到时间T为止的累计回馈。由于化学分子的可靠性只有在完整SMILES是才有效,因此我们将只评估完整序列的返回。
在给出状态下的一系列行动中获得收益,增强学习的目的及在于如何提高策略Agent下的累计回馈期望__E(G)。一个在step T 下具有明确终止点的任务被称为__episodic task,生成SMILES即是一episodic task。
用来训练agent的状态和行动既可以由agent自身生成,也可以由其他方式生成,分别称为__on-policy__和__off-polict__。off-policy和on-policy的根本区别在于off-policy学习的policy和agent实际执行的policy并不相同。
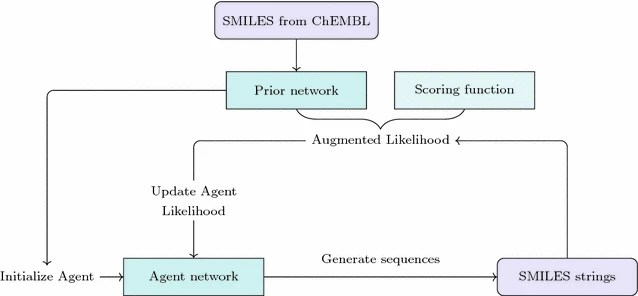
Figure 4. The Prior and Agent Structure
2.3. The Prior Network
极大似然估计用于训练由3层1024个GRU(forget bias 5)构成的初始RNN。训练集为RDkit生成的150万CHEMBL数据库中分子的canonical SMILES,这些分子被限制在包含10 - 50个重原子,且元素包含于{H, B, C, N, O, F, Si, P, S, Cl, Br, I}。模型使用最陡梯度下降法训练5000步,batch大小为128,使用Adam optimizer (β1 = 0.9, β2 = 0.9, ϵ = 10-8),一个初始learning rate为0.001和一个100步衰减的learning rate 0.02。梯度限制在[-3, 3],使用Tensoflow执行Prior以及增强学习Agent。
2.3. The Agent Network
我们限制将问题表示为通过一个部分观测的Markov决策过程的RNN,产生一个具有期望属性分子的SMILES。这一过程中,agent必须在给出当前cell时做出决定接下来选择哪一个字符character。我们使用Prior模型中学习到的条件概率分布作为先验策略。我们称使用先验策略prior policy的网络为___Prior___,修改后的网络为___Agent___。任务是episodic的,从RNN的第一步开始直到EOS token被采样。行动序列__A = a1, a2, …, aT__代表了这一eposid中产生的SMILES,而行动概率的积__P(A) = ∏Tt=1 π(at∣st)__代表了生成序列的概率。
另___S(A)___ <- __[-1, 1]__作为打分函数,粗糙地评价生成的SMILES序列是否含有期望的属性。所以现在就变成了从先验___πprior_更新agent的policy π,以达到提高生成序列的期望打分。但我们同时也想将新的policy与prior policy相锚定,绑定prior policy学习到的SMILES语法和CHEMBL数据库的化学结构分布。因此我们定义扩张的似然度 __ logP(A)𝕌:
logP(A)𝕌 = logP(A)Prior + σS(A)
其中σ是一个标量系数。因此序列A的回馈___G(A)__可以被视为Agent似然度__log P(A)A__和扩张似然度的协同:
G(A) = −[logP(A)𝕌 −logP(A)𝔸]2
因此Agent的目的即为学习一种可以最大化期望回馈的policy,即最小化损失函数___L(Θ) = -G___。Agent使用on-policy方法训练,在一个batch为128生成序列上,在每次生成batch和打分后更新π。使用learning rate为0.0005,梯度属于[-3, 3]的标准梯度下降法。
2.5 The DRD2 activity model
应用过程使用Dopamine D2 Receptor,DRD2作为靶标蛋白,相应的活性数据收集于ExCAPE-DB数据库。在这一数据集中,7218个活性化合物pIC50 > 5, 343204非活性化合物pIC50 < 5。随机取出100,000个非活性化合物子集。为了减少近邻相似性nearest neighbour similarity,活性分子产生Extended Connectivity Molecular Figerprint with diameter 6 (ECMFP6)指纹,使用RDKit的Butina聚类方法根据Tanimoto相似性进行聚类,cutoff为0.4。聚类中心分子被选出。这些聚类根据大小排序并逐个分给测试test,检验validation,训练集train,比例分别为1/6, 1/6, 4/6。与actives不超过0.5%的相似分子的inactives使用相同比例分配。
3. Results and Discussion
3.1. Structure generation by Prior
初始训练后,Prior生成的94%的序列是有效的分子结构,其中90%是区别于训练集的新结构。Figure 5显示了SMILES的生成,每一个token都存在一个Prior产生的条件概率分布。在这一例子中,O如果被采样,C、N和卤素均有可能被采出,-0.3 C,-2.7 N,-1.8 O,-5.0 F和Cl。
有几种观测现象:
- 若n被采样,会导致开环
- 若芳香环开环,芳香原子c,n,o,s变为probable,直到5,6步后闭环
- 学习到了多环结构的语法,一旦编码1被使用,新的环将会使用2
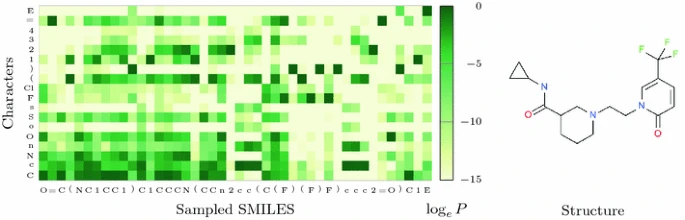
Figure 5. Conditional probability over the next token as a function of previously chosen ones according to the model.
3.2. Learning to avoid sulphur
为了验证Agent可以根据条件训练为不产生含硫S的分子,定义打分函数:
S(A) = 1, 0, −1
if valid and no S, if not valid, if contains S
Agent使用σ = 2训练1000步,并且使用Prior和Agent分别产生12800个分子就行比较,结果如Table 1。分子属性使用RDkit计算。
3.3 Similarity guided structure generation
下一个任务是能否根据query Structure产成相似结构。因此使用分子指纹的相似系数衍生的打分函数:
S(A) = −1 + 2 × min{Ji, j , k}/k
这定义了分子与query结构相似度k <- [0, 1]所对应的回馈为[-1, 1]。首先使用Celecoxib为query,采用较强的k = 1和sigma = 15条件下的采样,在200步后即生成了Celecoxib。
第二次使用了不含有Celecoxib与相似性大于0.5的分子的CHEMBL训练集,重新训练得到reduced Prior
,并与之前的canonical Prior进行比较。然后一个基于reduced Prior的Agent模型进行训练,在400步后,开始找到Celecoxib,在1000步后Celecoxib成为主要的结构产出,占比达到1/3。见Figure 7.
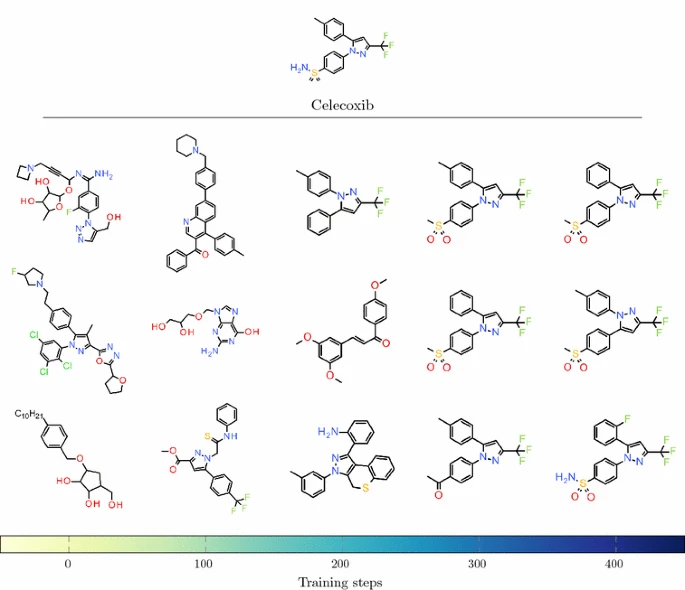
__Figure 7. Evolution of generated structures during training Structures sampled __
3.4 Target activity guided Structure generation
接下来使用具有生物活性的分子进行优化,即也是一种inverse QSAR的方法。如前所属,DRD2被作为标靶,活性的分子分别分为了1405,1287,4526三个集合。聚类减少了近邻相似性为0.53,随机为0.69.
打分函数变为:
S(A) = −1 + 2 × Pactive
这里貌似对Pactive没有解释,稍后阅读代码更新
Agents在sigma = 7的条件下训练3000步,Prior和对应的Agent分别产生128000条序列进行对比。训练后,预测的DRD2的活性化合物结构的占比由Prior的0.02提高到Agent的0.96 (Table 3)。测试集中的分子在两种Prior中的recover率为0%,在canonical Prior产生的Agent中为13%,reduced Prior产生的Agent中为7%。这表明我们可以产生__“novel”__的结构,并且既不属于DRD2活性模型也不属于Prior和实验验证的活化产物。
基于reduce Prior的Agent和基于canonical Prior的Agent在预测活性分子时具有相似效果,与已知的活性分子相似性却较低。
Figure 9显示出reduced Prior和对应的Agent的条件概率分布,可以看到增强学习后的概率分布并不会有剧烈的变化。但只要条件概率分布在某一步有一个较大的改变,就会对序列的极大似然度产生巨大的变化,从而改变生成的结构。
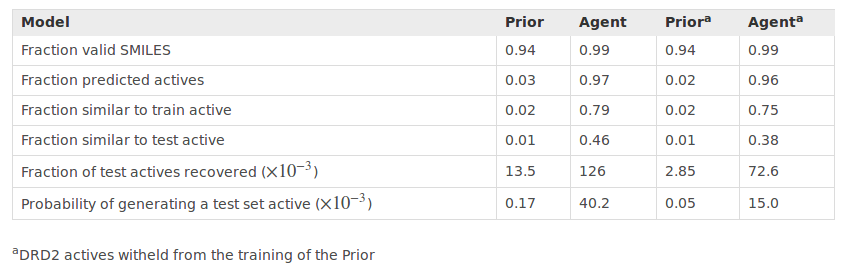
Table 3. Performance of the DRD2 activity model
4. Conclusion
- RNN是一种处理SMILES结构的有效方法。
- 对其他的RNN参数的调整值得进一步研究,也可以对token embedings方法进行增加。
- 以上的例子均使用了单变量打分函数,但实际上可以增加如目标活性,DMPK,合成可能性等多参数的打分函数。
读书笔记
其实文章只给了一些简单的例子,在应用过程中即便对RNN不甚了解,也可以尝试更改打分函数,来增加生成分子的限制。作者的github代码从TF/Python2.7更新为了PyTorch/Python3.6,希望自己有时间可以仔细阅读他的代码吧!
另外也有研究者开始使用GAN来达成相同目的,不过论文可读性还是这一篇更好学习些。
Reference
Olivecrona, M., Blaschke, T., Engkvist, O., & Chen, H. (2017). Molecular De Novo Design through Deep Reinforcement Learning. arXiv preprint arXiv:1704.07555.
Segler MHS, Kogej T, Tyrchan C, Waller MP (2017) Generating focussed molecule libraries for drug discovery with recurrent neural networks. arXiv:1701.01329
Hochreiter S, Schmidhuber J (1997) Long short-term memory. Neural Comput 9(8):1735–1780. doi:https://doi.org/10.1162/neco.1997.9.8.1735
Sun J, Jeliazkova N, Chupakin V, Golib-Dzib J-F, Engkvist O, Carlsson L, Wegner J, Ceulemans H, Georgiev I, Jeliazkov V, Kochev N, Ashby TJ, Chen H (2017) Excape-db: an integrated large scale dataset facilitating big data analysis in chemogenomics. J Cheminform 9(1):17. doi:https://doi.org/10.1186/s13321-017-0203-5